18 Oct Botrytis
Botrytis
Background
Erected by Micheli in 1729, the genus Botrytis is one of the first described genera of fungi. Persoon (1801) designated five species under the binomial system of Linnaeus validated the genus and included one of Micheli’s species, B. cinerea, so named by Von Haller (1771). The genus name refers to the structure of the macroconidia, which rise and form clusters with the shape of grape bunches: ‘botryose’. Botrytis is the asexual stage of Botryotinia. The Botrytis community has in its recent meeting (Italy, 23–28 June 2013) unanimously recommended the exclusive use of the asexual name Botrytis over Botryotinia, the name of the sexual stage, since Botrytis is historically the oldest name and it is commonly used by plant pathologists, breeders, and growers. In line with this recommendation, a list of generic names of fungi for protection under the International Code of Nomenclature has included this genus under the name Botrytis and not Botryotinia (Kirk et al. 2013). We, therefore, follow this recommendation in this paper and use Botrytis. Species of the genus Botrytis infect >250 host species, including major greenhouse and field crops such as tomato, grape, strawberry, onion, and ornamentals such as rose, lily, and tulip (Staats et al. 2005). Most Botrytis species are necrotrophic pathogens that (are able to) kill the host tissue during infection. Interestingly, an endophytic species (B. deweyae) has recently been discovered, which under appropriate conditions can cause ‘spring sickness’ in ornamental Hemerocallis (daylily) hybrids (Grant- Downton et al. 2014). Botrytis cinerea is the best-studied species in the genus (Williamson et al. 2007) and was recently elected as the second most important plant pathogenic fungal species (Dean et al. 2012).
In the asexual state, Botrytis produces different tissues including mycelia, macroconidia, microconidia, and sclerotia. Macroconidia are ellipsoidal to obovoid shape and rise from conidiophore branches into botryose clusters. They are pale brown and range in size from 9–23×8–15 μm. Microconidia are more spherical and much smaller than macroconidia (about 1 μm), and function as male spermatia (Groves and Loveland 1953; Faretra et al. 1988; Beever and Parkes 1993; Fukumori et al. 2004). Sclerotia are irregularly hemispherical, convex and normally have a concave surface. They are usually black, with sizes ranging between 1 and 10 mm (Whetzel 1945), and function as survival structures during winter and serve as a maternal parent in the production of apothecia.
The sexual state forms fruiting bodies called apothecia: a cup- or open saucer-shaped ascoma at the top of a stalk, that acts as a platform to discharge ascospores from the ascus. Botrytis apothecia vary in size depending on the species, between 1 and 25 mm high and 1–6 mm diam. (Hennebert and Groves 1963; Bergquist and Lorbeer 1972). Apothecia are brown and become darker when mature (Hennebert and Groves 1963; Bergquist and Lorbeer 1972; Faretra and Antonacci 1987). Generally, multiple apothecia can develop on a single sclerotium. Mature apothecia normally can be observed 2 months after fertilization (Faretra et al. 1988; Hennebert and Groves 1963; Van Der Vlugt-Bergmans et al. 1993). In the genus Botrytis, both homothallic and heterothallic reproductive lifestyles have been reported. Homothallic (self- fertile) species can undergo sexual reproduction and form apothecia and generate progeny in the absence of a mating partner, e.g. B. porri and B. globosa (Buchwald 1953; Elliott 1964). By contrast, heterothallic (self-sterile, self-incompatible) species require isolates with compatible mating types in order to complete the sexual cycle. B. cinerea is considered a typical heterothallic fungus (Elliott 1964; Faretra et al. 1988). Mating is controlled by the mating-type locus with two alleles, MAT1-1, and MAT1-2 (Faretra et al. 1988), each carrying two distinct, non-homologous genes (Amselem et al. 2011).
Species identification and numbers
Approximately half of the Botrytis species are named after the host that they are derived from (listed in Table). One hybrid species, B. allii which originated from hybridization between B. byssoidea and B. aclada (Nielsen and Yohalem 2001; Yohalem et al. 2003) could not be placed in the phylogeny (Staats et al. 2005) and was omitted from Table 3. The genus Botrytis predominantly comprises narrow host range pathogens that infect a single, or a few (often related) host species. There are two exceptions to this rule: B. cinerea can infect more than 200 host species (Jarvis 1977 ), and B. pseudocinerea has been isolated from several unrelated host species (Fournier et al. 2005; Leroch et al. 2013).
The taxonomic classification and nomenclature in Botrytis have rarely been comprehensively reviewed. Morphological descriptions of most species have been published in the 19th and the first half of the 20th century in separate papers, many of which are not easily accessible. The most recent taxonomic compilation of the genus is in a monograph by Jarvis (1977), which also lists ~25 excluded or doubtful species, and briefly describes the historical debates between mycologists and the confusion in the classification of Botrytis species. Morphological features were often inadequate to distinguish species and the variability among isolates of the same species further complicated the situation (Jarvis 1977). Recent studies have identified B. cinerea and B. pseudocinerea as species that are very similar in morphology, yet recognized as distinct taxa that diverged several million years ago (Walker et al. 2011). Even more puzzling, the morphology and narrow host range of B. fabae separate this species clearly from B. cinerea and B. pseudocinerea, but phylogenetic studies revealed it to be a sister species of B. cinerea (see below). These examples illustrate the limitations of morphological characters for Botrytis species identification.
Table Botrytis. Details of the isolates used in the phylogenetic tree
| Species | Isolate | Host | GenBank accession numbers | ||||
| RPB2 | HSP60 | G3DPDH | NEP1 | NEP2 | |||
| Botrytis aclada | MUCL8415 | Allium spp. | AJ745664 | AJ716050 | AJ704992 | AM087059 | AM087087 |
| B. byssoidea | MUCL94 | Allium spp. | AJ745670 | AJ716059 | AJ704998 | AM087045 | AM087079 |
| B. calthae | MUCL1089 | Caltha palustris | AJ745672 | AJ716061 | AJ705000 | AM087031a AM087088a | |
| B. cinerea | MUCL87 | >200 species | AJ745676 | AJ716065 | AJ705004 | DQ211824a | DQ211825a |
| B. caroliniana | CB15* | Rubus fruticosus | JF811590 | JF811587 | JF811584 | JF811593 | NA |
| B. convoluta | MUCL11595 | Iris spp. | AJ745680 | AJ716069 | AJ705008 | AM087035 | AM087062 |
| B. croci | MUCL436 | Crocus spp. | AJ745681 | AJ716070 | AJ705009 | AM087047 | AM087065 |
| B. deweyae | CBS134649* | Hemerocallis spp. | HG799518 | HG799519 | HG799521 | HG799527 | HG799520 |
| B. elliptica | BE9714 | Lilium spp. | AJ745684 | AJ716073 | AJ705012 | AM087049 | AM087080 |
| B. fabae | MUCL98 | Vicia spp. | AJ745686 | AJ716075 | AJ705014 | DQ211829 | DQ211831 |
| B. ficariarum | MUCL376 | Ficaria verna | AJ745688 | AJ716077 | AJ705016 | AM087056 | AM087085a |
| B. fabiopsis | BC-2* | Vicia faba | EU514473 | EU514482 | EU519211 | NA | NA |
| B. galanthina | MUCL435 | Galanthus spp. | AJ745689 | AJ716079 | AJ705018 | AM087057 | AM087067a |
| B. gladiolorum | MUCL3865 | Gladiolus spp. | AJ745692 | AJ716081 | AJ705020 | AM087041 | AM087072a |
| B. globosa | MUCL444 | Allium ursinum | AJ745693 | AJ716083 | AJ705022 | AM087044a AM087071 | |
| B. hyacinthi | MUCL442 | Hyacinthus spp. | AJ745696 | AJ716085 | AJ705024 | AM087048 | AM087066a |
| B. narcissicola | MUCL2120 | Narcissus spp. | AJ745697 | AJ716087 | AJ705026 | AM087046 | AM087078 |
| B. paeoniae | MUCL16084 | Paeonia spp. | AJ745700 | AJ716089 | AJ705028 | AM087033 | AM087064a |
| B. pelargonii | CBS 497.50 | Pelargonium spp. | AJ745662 | AJ716046 | AJ704990 | AM087030 | DQ211834a |
| B. polyblastis | CBS287.38 | Narcissus spp. | AJ745702 | AJ716091 | AJ705030 | AM087039 | AM087074 |
| B. porri | MUCL3234 | Allium spp. | AJ745704 | AJ716093 | AJ705032 | AM087060 | AM087063 |
| B. pseudocinerea | VD110 | Vitis vinifera | Unpublished | Unpublished | Unpublished | NA | NA |
| B. ranunculi | CBS178.63 | Ranunculus spp. | AJ745706 | AJ716095 | AJ705034 | AM087054 | AM087086 |
| B. sinoallii | HMAS250008 | Allium spp. | EU514479 | EU514488 | EU519217 | NA | NA |
| B. sphaerosperma | MUCL21481 | Allium triquetrum | AJ745708 | AJ716096 | AJ705035 | AM087042 | AM087068 |
| B. squamosa | MUCL1107 | Allium cepa | AJ745710 | AJ716098 | AJ705037 | AM087052 | AM087084 |
| B. tulipae | BT9830 | Tulipa spp. | AJ745713 | AJ716102 | AJ705041 | AM087037 | AM087077 |
| Monilinia fructigena | 9201 | Stone fruit and pome fruit | AJ745715 | AJ716047 | AJ705043 | NA | NA |
| Sclerotinia sclerotiorum | 484 | >400 species | AJ745716 | AJ716048 | AJ705044 | NA | NA |
Ex-type (ex-epitype) strains are bolded and marked with an * and voucher strains are bolded
a sequences obtained from a different isolate than the one listed in the table.
Molecular phylogeny
Holst-Jensen et al. (1998) were the first to use nuclear ribosomal ITS sequences to infer a phylogeny of the family Sclerotiniaceae, including several members of the genus Botrytis. The relationships among many Botrytis species could not be resolved because of the limited number of informative characters, however, the study permitted the conclusion that Botryotinia asexual morphs along with Botrytis sexual morphs constitute a monophyletic lineage (Holst-Jensen et al. 1998). The phylogeny of the Sclerotiniaceae was further refined by Andrew et al. (2012) using three protein-coding genes: calmodulin, glyceraldehyde 3-phosphate dehydrogenase G3PDH, and heat shock protein HSP60.
Staats et al. (2005) performed a comprehensive phylogenetic analysis of the genus Botrytis, at that time comprising 22 recognized species and one hybrid. Using three protein-coding genes (G3PDH, HSP60, and the DNA-dependent RNA polymerase subunit II gene RPB2), they corroborated the morphological and host plant-based classification of Botrytis spp. and divided the genus into two (rather widely separated) clades. Clade I contained species that only infect eudicot plants, while Clade II contained species that can infect either eudicotyledonous or monocotyledonous plants. The use of the same three genes facilitated the discovery of Botrytis sinoallii, a new species infecting Allium spp., and its distinction from other Botrytis spp. infecting the same hosts (Zhang et al. 2010b); B. fabiopsis, a new species infecting broad bean, very distant from B. fabae (Zhang et al. 2010a); and B. caroliniana, a new species infecting blackberry (Li et al. 2012).
Two genes, encoding phytotoxic proteins NEP1 and NEP2, were shown to provide higher resolution in distinguishing species in the genus Botrytis because they seem to be the subject of higher evolutionary rates than the housekeeping genes G3PDH, HSP60 and RPB2 (Staats et al. 2007a). The NEP1 and NEP2 genes were shown to have evolved under positive selection which suggested the role of these proteins in the infection process (Staats et al. 2007a). One might, therefore, infer that such genes cannot serve as neutral phylogenetic markers. Functional analysis in B. cinerea and B. elliptica using targeted knockout mutants failed to reveal a role of NEP genes in virulence of these two species (Staats et al. 2007b; Cuesta Arenas et al. 2010), which would lend support to considering these genes as neutral markers and adequate tools in phylogeny.
The studies by Staats et al. (2005) revealed incongruence between the phylogenies of Botrytis spp. and their hosts. Species infecting the same host clustered in different (sub) clades, e.g. B. aclada, B. squamosa, B. porri, B. byssoidea and B. sinoallii all infecting Allium. Conversely, closely related species can infect very different hosts, e.g. B. elliptica infecting the monocotyledonous host Lilium and B. ficariarum infecting the dicotyledonous host Ficaria (Staats et al. 2005). More recently, similar incongruence has been reported for newly described species, e.g. B. fabiopsis infecting Vicia faba is very distant from B. fabae infecting the same host (Zhang et al. 2010a), and B. caroliniana infecting blackberries and strawberries are very distant from B. cinerea (Li et al. 2012).
Recently, Khan et al. (2013) combined data from ITS and IGS regions with the G3PHD gene, with the aim of improving molecular identification of Botrytis species that cause neck rot disease on the onion. ITS and IGS regions were insufficiently informative to distinguish B. allii and B. byssoidea. The sequences of ITS and IGS for B. allii and B. byssoidea confirmed that they have a close relationship, but G3PDH sequences of several B. allii isolates were clearly distinct, some clustering with B. aclada and others clustering with B. byssoidea (Khan et al. 2013), as might be expected for a hybrid species.
Sequence analysis of the G3PDH and β-tubulin genes amplified from herbarium specimens of Botrytis collected from grey mold-infected apple (deposited in 1932) enabled O’Gorman et al. (2008) to corroborate the existence of B. mali, a species that had been published (Ruehle 1931), but by lack of description was considered doubtful.
The figure shows a maximum likelihood tree of Botrytis spp., based on concatenated sequences of parts of the three genes G3PDH, HSP60 and RPB2 (amplified using primers defined by Staats et al. (2005). Five species described after the publication of the phylogeny by Staats et al. (2005), i.e. B. caroliniana, B. deweyae, B. fabiopsis, B. pseudocinerea and B. sinoallii, clearly cluster within the genus and are genuine Botrytis species. Botrytis mali could not be included in the tree due to a lack of sequences for the HSP60 and RPB2 genes. Based on G3PDH and ß-tubulin sequences it would cluster with B. paeoniae (O’Gorman et al. 2008).
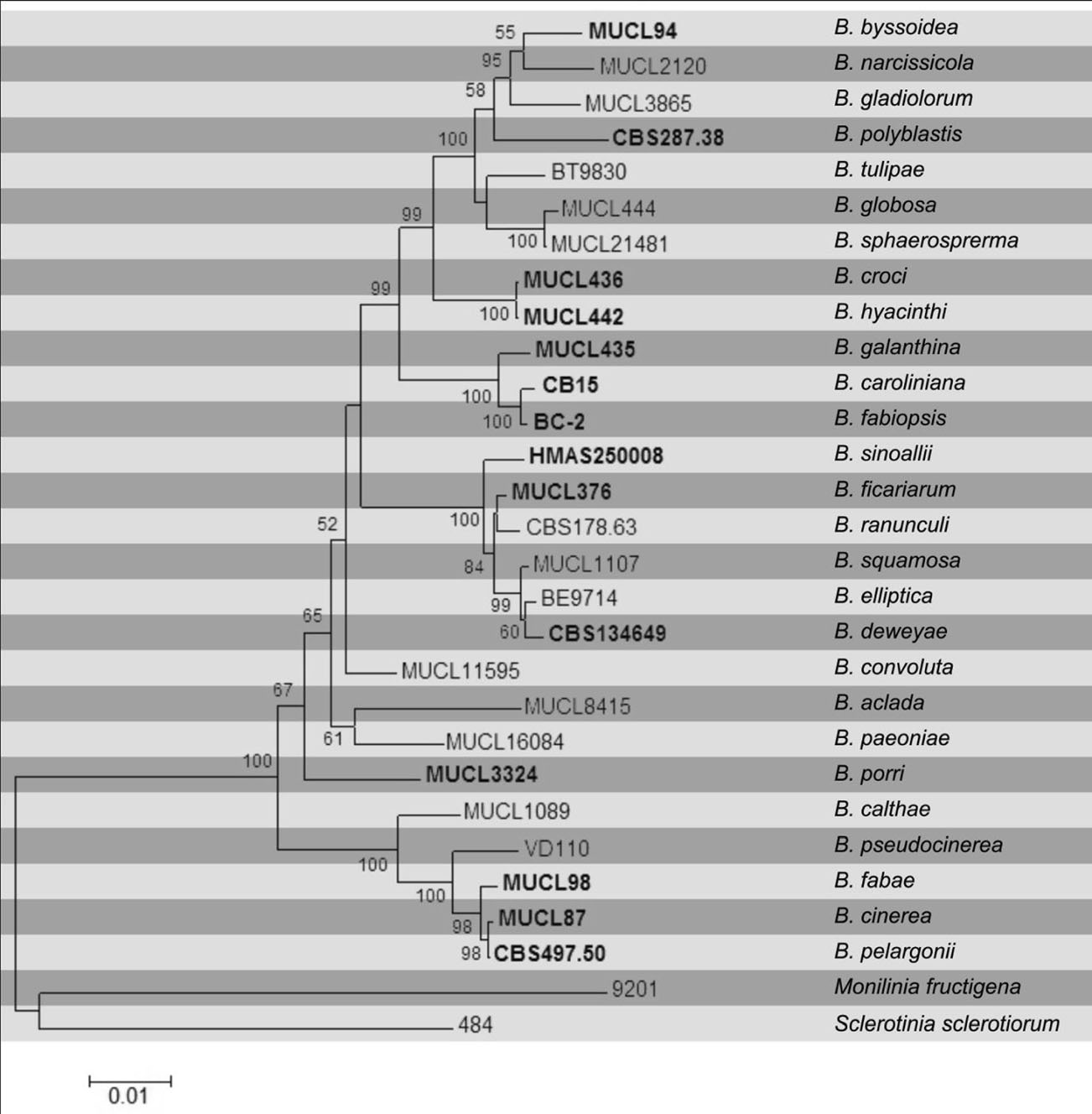
Fig. Phylogramgenerated from Maximum likelihood analysis based on combined sequences of G3PDH, HSP60 and RPB2 from 28 recognized Botrytis species. Bootstrap support values greater than 50 % are indicated above/below the nodes. The ex-type (ex-epitype) and voucher strains are in bold. The tree is rooted with Monilinia fructigena and Sclerotinia sclerotiorum
The Botrytis cinerea species complex
The Botrytis ‘dicot’ clade I consist of B. cinerea, B. pelargonii, B. fabae, B. pseudocinerea and B. calthae. Molecular data do not fully support a separation between B. pelargonii and B. cinerea (Staats et al. 2005, 2007a; Plesken et al. 2014), and the existence of B. pelargonii as a separate species is therefore doubtful. As mentioned above, B. cinerea and B. pseudocinerea are morphologically very similar yet phylogenetically more distant from each other than B. cinerea and B. fabae. All genes tested so far place B. calthae as most remote to all other clades I species.
Botrytis cinerea not only has a broad host range but also shows considerable phenotypic variability in vegetative growth, conidiation and sclerotium formation (Kerssies et al. 1997; Martinez et al. 2003; Schumacher et al. 2013). Numerous studies have documented a similar variability in genotypic characters, such as amplified restriction length polymorphism, detection of transposable elements and microsatellite heterogeneity. Recently, B. cinerea strains have been described that produce bikaverin, a reddish pigment. These strains contain an intact bikaverin biosynthesis gene cluster (presumably acquired by horizontal gene transfer from Fusarium), which is partially deleted and nonfunctional in most non-bikaverin producing B. cinerea strains (Campbell et al. 2012; Schumacher et al. 2013)
A subdivision of B. cinerea into genetically distinct groups has proved to be difficult. Analysis of the presence or absence of two types of transposable elements, named Boty (Diolez et al. 1995) and Flipper (Levis et al. 1997), was adopted as a tool to divide isolates into four transposon types, Transposa (isolates having both elements), Vacuma (isolates having neither element), Boty and Flipper (Giraud et al. 1997, 1999). This classification led to the discovery of B. pseudocinerea, which is usually Vacuma, but the transposon-based classification turned out to be of limited use since B. cinerea populations appear to consist of mixtures of different transposon types. Intriguingly, the predominance of a certain type appears to be influenced by the host. While on grapes, strawberries, and tomatoes, Transposa types are predominant, whereas B. cinerea populations from kiwi and apples are dominated by Vacuma types (Esterio et al. 2011; Johnston et al. 2013; Muñoz et al. 2002; Samuel et al. 2012; M. Hahn, unpublished). The reasons for this observation are unknown.
Evidence for genetic differentiation of B. cinerea populations with different host preferences was obtained with microsatellite markers. In France, isolates from grapes and blackberries were shown to be divergent, indicating limited gene flow between populations on these host plants (Fournier and Giraud 2008). A recent study on grey mold isolates from fungicide-treated strawberry fields revealed the existence of a predominant B. cinerea genotype, named group S, that is closely related to but distinct from the common genotype of B. cinerea (Leroch et al. 2013). Sequencing of the highly polymorphic MRR1 gene revealed that group S isolates show more than 4 % divergence from B. cinerea strains B05.10 and T4, which have MRR1 genes with 99.9 % identity. Further sequencing of HSP60 and NEP2, and of two FUNYBASE genes that are suitable for phylogenetic studies (Marthey et al. 2008), partially supported the genetic separation of group S isolates (Johnston et al. 2013; Leroch et al. 2013). Genome sequencing of several B. cinerea and group S strains and the analysis of additional polymorphic genes in isolates collected from various host plants in different countries revealed at least two subclades that could be separated from the common B. cinerea genotype (Plesken and Hahn, unpublished). In fungicide-treated strawberry fields group S isolates dominated, whereas grapes were infected almost exclusively by common B. cinerea genotypes. These data, together with those of putative new endophytic Botrytis taxa that grouped close to B. cinerea (Shipunov et al. 2008), support the idea that B. cinerea represents a species complex, comprising genetically and phenotypically distinct groups.
Recommended genetic markers
G3PDH, RPB2 and HSP60—placement within the Sclerotiniaceae and the ascomycetes
NEP1 and NEP2—for higher resolution within the genus Botrytis,
The NEP1 and NEP2 genes are under positive selection (Staats et al. 2007a) and potentially influence interactions with the host plants. The NEP genes should, therefore, be used with caution.
Research is ongoing to identify a set of highly polymorphic genes that better resolve the phylogeny of taxa in clade I (Hahn et al., unpublished). It remains to be established whether those genes are equally useful for resolving the clade II species and whether universal primers can be designed before these genes can be employed to infer a comprehensive phylogeny of the entire genus.
No Comments